Research Topics
1. Excited-State Dynamics in Optoelectronic Molecular Materials
The nonequilibrium excited-state dynamics in organic conjugated
molecules, molecular aggregates, and molecular crystals are
crucial for their optoelectronic performance. Our research
focuses on understanding the microscopic mechanisms of
electronic processes, including energy transfer, transport,
separation, and relaxation. By applying tensor-network-based
quantum dynamics methods, we aim to reveal how electronic
structure and electron–vibration coupling influence these
dynamics, thereby bridging theoretical predictions with emerging
experimental observations in molecular optoelectronics. Recently,
we found that excitonic coupling can greatly influence the
nonradiative decay rate in molecular aggregates, leading to an
apparent "violation" of the classical energy gap law [1-2].
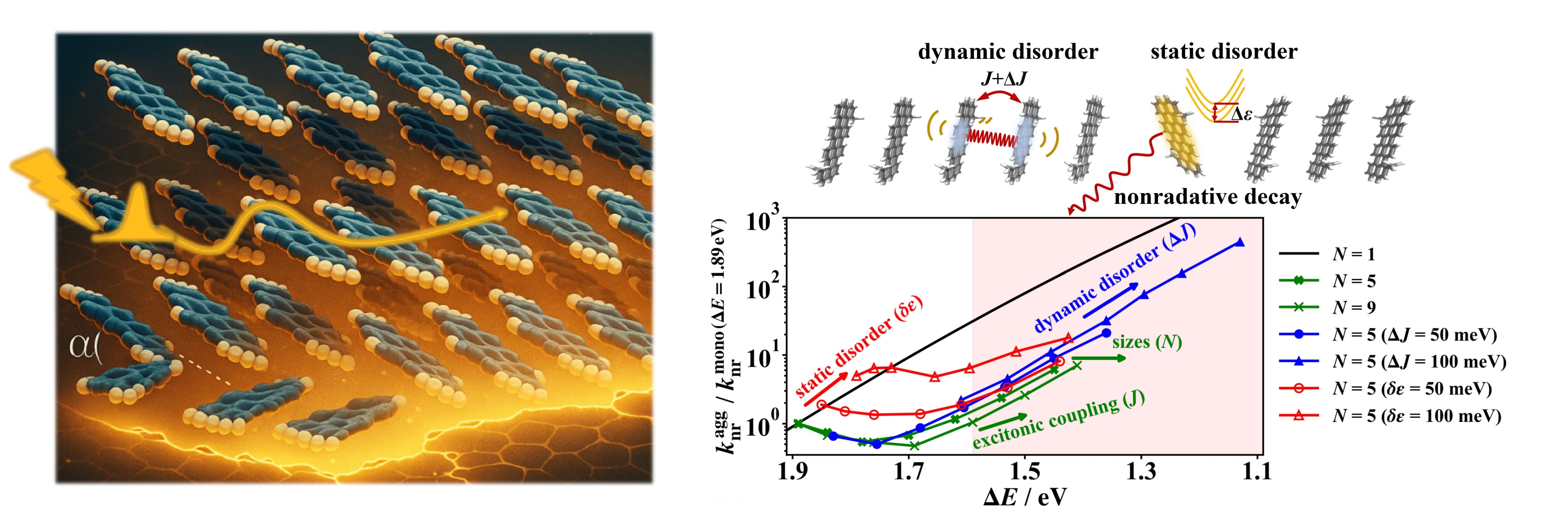
[1] Yuanheng Wang; Jiajun Ren*; Zhigang Shuai*. Nature Communications 2023, 14 (1), 5056.
[2] Zhao Zhang; Yijia Wang; Xiaoyan Zheng; Jiajun Ren*; Zhigang
Shuai; Wei-Hai Fang. The Journal of Physical Chemistry C 2025, 129 (27), 12520-12530.
2. Tensor-Network-Based Methods for Quantum Dynamics in Complex Systems
Exact simulation of high-dimensional quantum dynamics faces the
quantum exponential wall, where computational cost scales
exponentially with system size. To overcome this, we develop
highly accurate and efficient tensor-network algorithms,
particularly the time-dependent density matrix renormalization
group (TD-DMRG) method, incorporating optimized operator
factorization and time-evolution schemes. These methods enable
the scalable and accurate treatment of electron–vibration
coupled dynamics, providing powerful tools for photophysical
processes in complex environments [3]. We recently developed a
hybrid method that integrates tensor-network algorithms with the
real-time path integral approach for simulating dissipative
open-system dynamics [4].
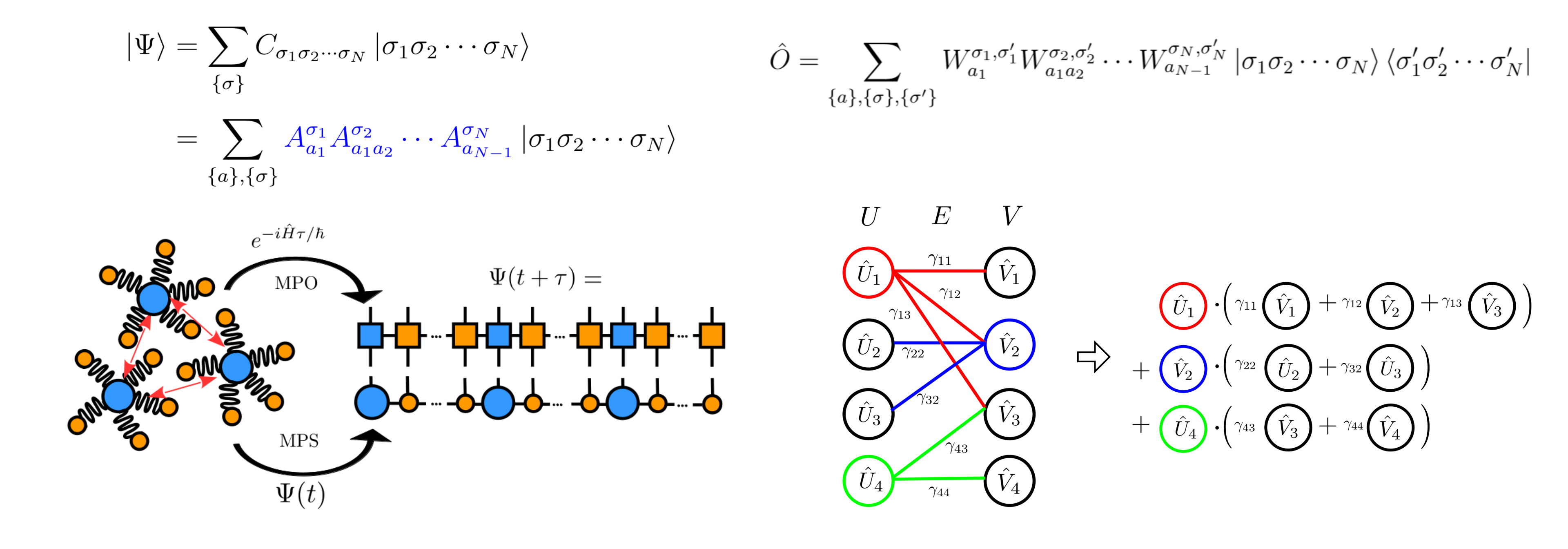
[3] Jiajun Ren*; Weitang Li; Tong Jiang; Yuanheng Wang; Zhigang
Shuai*. Wiley Interdisciplinary Reviews: Computational Molecular
Science 2022, 12 (6), e1614.
[4] Limin Liu; Jiajun Ren*; Wei-Hai Fang. The Journal of
Chemical Physics 2024, 161 (8), 084101.
3. Quantum Algorithms for Quantum Dynamics
Simulating quantum dynamics on quantum devices is an exciting
frontier, yet current algorithms for closed and open quantum
systems remain challenging. Furthermore, today’s hardware is
limited by noise. Our goal is twofold: (1) to design quantum
dynamics algorithms with genuine quantum advantage for future
fault-tolerant quantum computers; and (2) to develop hybrid
quantum–classical approaches that extract meaningful results on
noisy intermediate-scale quantum (NISQ) devices. We are also
exploring quantum-inspired classical algorithms and classically-inspired
quantum algorithms, leveraging insights from both paradigms.
Recently, inspired by the multi-set wavefunction formalism used
in classical tensor network algorithms, we have proposed a
multi-set variational quantum dynamics algorithm that extends
quantum simulations to nonadiabatic problems [5].
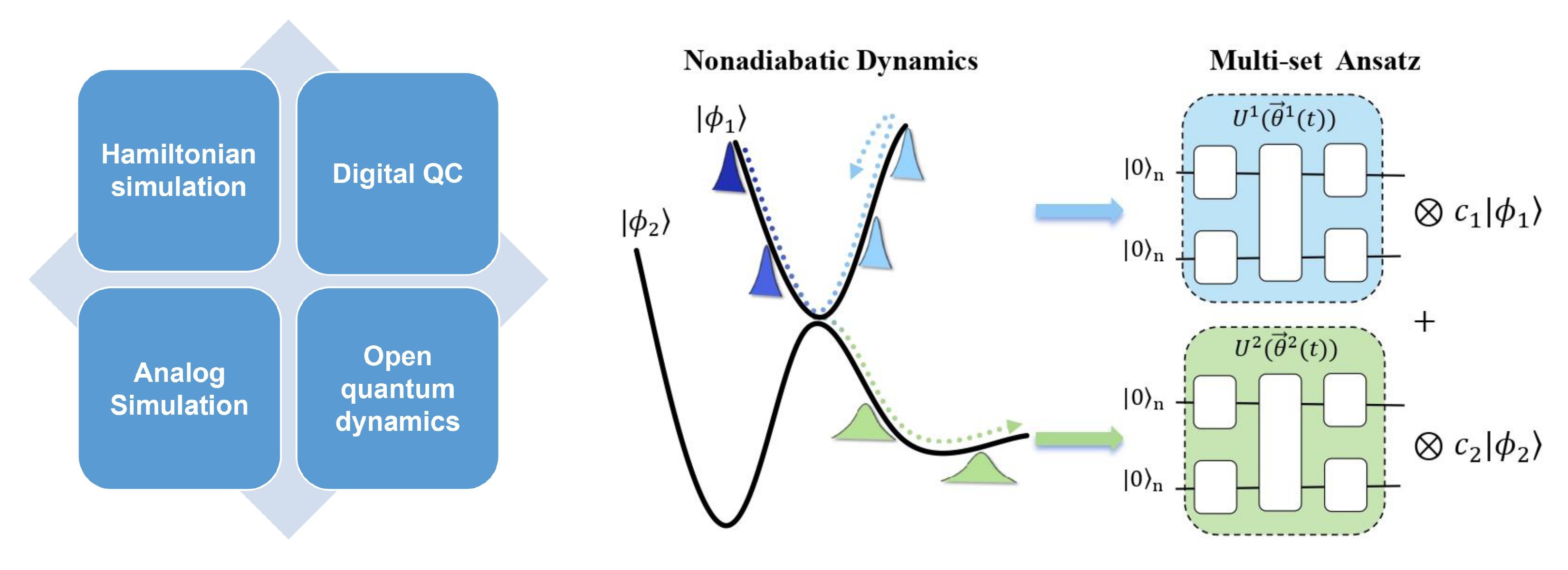
[5] Jingjing Li; Weitang Li; Xiaoxiao Xiao; Limin Liu; Zhendong Li; Jiajun Ren*; Wei-Hai Fang. The Journal of Physical Chemistry Letters 2025, 16 (16), 3911-3919.